Gaucher S Disease
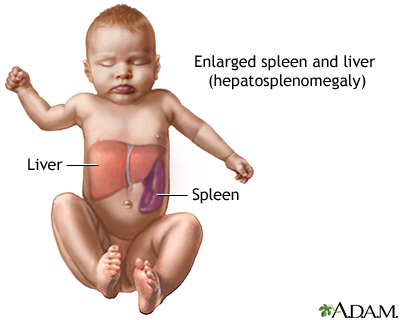
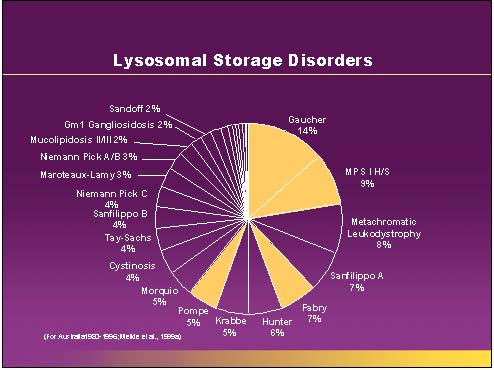
Gaucher's disease ( /ɡoʊˈʃeɪz/) is a genetic disease in which a fatty substance (lipid) accumulates in cells and certain organs. Gaucher's disease is the most common of the lysosomal storage diseases. It is a form of sphingolipidosis (a subgroup of lysosomal storage diseases), as it involves dysfunctional metabolism of sphingolipids. The disorder is characterized by bruising, fatigue, anemia, low blood platelets, and enlargement of the liver and spleen. It is caused by a hereditary deficiency of the enzyme glucosylceramidase. The enzyme acts on the fatty acid glucosylceramide. When the enzyme is defective, glucosylceramide accumulates, particularly in white blood cells, most often macrophages (mononuclear leukocytes). Glucosylceramidase can collect in the spleen, liver, kidneys, lungs, brain and bone marrow. Symptoms may include enlarged spleen and liver, liver malfunction, skeletal disorders and bone lesions that may be painful, severe neurologic complications, swelling of lymph nodes and (occasionally) adjacent joints, distended abdomen, a brownish tint to the skin, anemia, low blood platelets and yellow fatty deposits on the white of the eye (sclera). Persons affected most seriously may also be more susceptible to infection. Some forms of Gaucher's disease may be treated with enzyme replacement therapy. The disease is caused by a recessive mutation in a gene located on chromosome 1 and affects both males and females. About 1 in 100 people in the United States are carriers of the most common type of Gaucher disease.
Gaucher S Disease
Gaucher S Disease

Gaucher S Disease

Gaucher S Disease
Gaucher S Disease

Gaucher S Disease

Gaucher S Disease

Gaucher S Disease

No comments:
Post a Comment